
MECHANICAL TRIGGERS TO PROGRAMM CELL DEATH IN
and how to prevent their Action in Failing Hearths
Proposed research within the context of current knowledge.
Cardiovascular diseases (CVDs) represent ~30% of the worldwide mortality (World Health Organization). Heart failure (HF) is a main form of CVD that occurs due to coronary heart disease, increased workload, or genetic predisposition. All types of HF share a progressive loss of cardiac myocytes by cell death (apoptosis), which is a dominant cause of disease progression. Understanding the mechanisms of cardiac apoptosis is the key to deciphering the molecular basis of the pathology and to develop viable diagnostics and therapeutic interventions in patients. To date, knowledge of myocardial cell death has remained fragmented and incomplete. This is largely due to the multiple pathological events that are involved, which include apoptosis, autophagy, necrosis, necroptosis (programmed necrosis, (see1.-3.)), and loss of cytoskeletal filaments - together these processes interlink biomechanical stress and cell survival.(4) Recently, the term mechanoptosis has been coined to describe such novel interlinks, i.e. the complex cell death mechanisms that emerge from the cross-talk between the mechanical stress of the sarcomeres (the contractile units of the cardiac myocytes) and the regulation of gene expression in the cell nucleus.(5)
Data by the participating TNE teams indicate that mechanoptotic signaling emanates from the giant elastic titin filament in the myocardium (Fig. 1). Among other recent findings, we have demonstrated negative myocardial apoptotic signalling of Tcap by its interaction with p53 (6), an axis that is in vivo down-regulated by MuRF1 and MuRF2.(7) We have also detected cardiomyocyte protection from apoptosis after MuRF1,2 inactivation (see below), and up- regulation of pro-apoptotic MuRF1, MuRF2, CARP, and FHL1 after titin stiffening.(8)Conversely, we noted down-regulation of stress atrogenes after titin relaxation in an RBM20 deficient background (see below), consistent with our earlier observation that isoform shifts to more compliant titins in patients with HF correlated with a better prognosis.(9) Finally, recent work (Labeit, in preparation) indicates that small molecules that target the titin kinase-MuRF1 M-band signalsome provide relieve from apoptosis-induced contractile dysfunction.
Importantly, the identification of these specific, targetable, cellular processes point to achievable therapeutic avenues for attenuating cardiomyocyte loss during HF. To develop such therapies, we urgently need to enhance our understanding of how myocardial programmed cell death is promoted by the biomechanical overload of the sarcomere. To achieve this within this network, we will first elucidate the process of sarcomere mechanosensation as mediated by the titin filament and the titin-based signalosomes (Fig. 2) that regulate apoptosis in HF. Then, we will develop the tools to down-regulate their involvement in stress-activation in intact hearts, contributing to unlocking their therapeutic potential for treating HF.
Originality. The importance of cell slippage and apoptosis in HF has been well recognized in acute infarcts, where apoptosis is considered secondary to ischemia, calcium-overload, and end-stage proteolysis.(10) Our novel mechanistic molecular hypothesis is that cell loss is an active process driven by the titin filament that integrates stretch signals with cell survival. Our findings strongly indicate that a vicious cycle of deranged sarcomeric elasticity and defective responses of the stretch-sensing signaling nodes that converge on titin is a leading primary patho-mechanism in HF. In support of this concept, a recent genetic study (11) identified a mutation in titin’s spring element as causative for arrythmogenic cardiomyopathy (AC), a heart muscle disease caused by a primary breakdown of healthy myocardium. Single molecule force spectroscopy showed that the titin mutation at physiological force levels triggered local unfolding. (12)
We postulate that titin chain regions so disrupted create rafts with increased viscoelasticity (“sticky“ segments) that alter the stretch response of titin’s molecular spring region, resulting in pathological tension levels in the myocardium. Structurally labile segments of titin would finally undergo proteolysis, leading to a pathological redistribution of signalosome interactions in the sarcomere, including disruption of communication shuttles between the sarcomere and the cell nucleus, further aggravating mechanosensory signalling. Such a scenario can explain how relatively small and common defects in the sarcomere lattice (such as point mutations in the titin chain that are abundant in humans, see below), in combination with prolonged stress, progressively amplify the effect and eventually result in a severe cardiomyocyte loss: The heart is exposed to life-long stretch-release cycles, and has low regenerative potential, making titin-mediated mechanoptosis a likely key pathomechanism in HF
Significance of the Project.
In developed countries, around 2% of adults suffer from heart failure, and in those over the age of 65, the incidence is 6–10%. The changing demographics in industrialized countries together with cardiac risk factors derived from life style, metabolic diseases, or a familial pre- disposition is about to transform HF into a major epidemic of modern societies, where progressive cardiomyocyte loss is a common end-stage feature. This project brings together a comprehensive and interdisciplinary team of world-leading experts who can identify the cellular mechanisms underlying progressive cytoskeletal pathologies in HF and unlock therapeutic potentials residing in the involved signalosomes. Research findings from this network will also support the development of innovative regenerative/protective therapies such as those that apply cardiac stem-cells/ iPS-cells: Regenerative cell applications will be more effective if endogenous mechanoptosis can be regulated. Thus, we anticipate that the impact of our findings on the clinical and research community will be far reaching and will eventually translate into improvements in the medical management of HF.
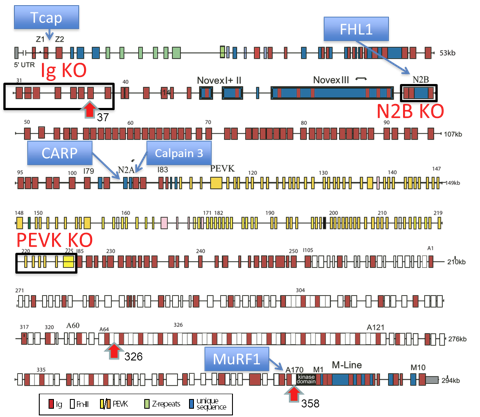
Figure 1. Overview of the 363 exons in the titin gene(13).
and stress sensing titin-binding proteins (blue boxes). Tcap binds to Z-disc titin and links in the nucleus to apoptosis and transcriptional regulation, CARP and calpain-3 provide a novel link between the titin spring region and the FAS apoptotic pathway via SIVA2, the titin kinase links to stress sensing via MURF1. In this TNE, KO mouse models are already available for Tcap, CARP, calpain-3, FHL1&2, MuRF1&2, and for titin deletion models (black boxes). Red arrowheads indicate locations of titin mutations that have been introduced into mice and represent novel mouse models for AC (exon 37), and DCM (exons 326 and 358; see text for details).
Specific aims to address our overall objectives. In order to (a) establish the mechanisms leading to mechanoptotic heart failure, and (b) develop therapeutic interventions that attenuate this condition, we will address four major research targets:
- Mechanical failure of the titin cytoskeleton as trigger of cell death. We will examine mechanoptotic activity in mouse models that have genetically altered titin compliances. In parallel we will study biopsies from HF patients with normal systolic function but increased diastolic stiffness and study titin-based stiffness and titin-based signalling.
- Pathological alteration of cytoskeleton-based signalling nodes. Our preliminary studies indicate that pathological titin tension ultimately deregulates signalling pathways in charge of cardiomyocyte health. The applicants will identify critical players, and their cross-connections to titin. The determination of 3D biosensing structures that can underpin the development of small molecule inhibitors will also be pursued.
- Failure of communication channels between the mechanosensory cytoskeletal nodes and other cell organelles. Pathological stress induces a deregulation of the feedback loops connecting the sarcomere with nuclear transcription, mitochondrial metabolism, and cytoplasmic autophagosomes. We will determine the key molecular events involved in these cellular communications and their alterations in HF.
- Translational avenues – containment of failing springs and pathological stress signalling in the sarcomere. Based on insights gained in Aim 1-3, the applicants will perturb key genes and key recognition interfaces in sarcomeric titin-based stress sensing signalosomes, as well as stabilize structurally labile spring components of titin. The goal is to attenuate mechanoptosis in the failing heart.
Bibliography (joint TNE publications in bold).
 Prof. Siegfried Labeit
Prof. Siegfried Labeit
 Fondation Leducq
Fondation Leducq